The Weakened ‘Glue’ In Your Body
Marfan syndrome is a genetic disorder that affects connective tissues in the body. In 75% of cases, Marfan syndrome is inherited from an affected parent in an autosomal dominant fashion. This leads to an abnormal production of fibrillin, a protein that is fundamental for the formation of elastic fibres found in connective tissues 1. In the remaining 25% of cases, the fibrillin-1 gene (FBN1) mutates sporadically in the parent’s egg or sperm — neither parent has Marfan syndrome, but the child will develop the syndrome due to the mutated gene in the egg/sperm.
Connective tissues, which surround body organs, are made up of fibres. They act like a ‘glue’ that connects, supports, and maintains body structure and other tissues or organs in the body. Individuals with Marfan syndrome are often tall and slim, presenting with abnormally long fingers and limbs, and have arm length that exceeds their body height. The two principal features of Marfan syndrome are lens dislocation and heart defects. Other common complications include craniofacial abnormalities, scoliosis/kyphosis, sunken or protruding chest, fatigue, and shortness of breath 2.
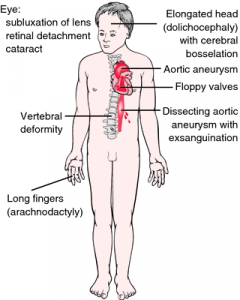
Common features of Marfan syndrome
Image credit: http://medical-dictionary.thefreedictionary.com/Marfan+syndrome
The most common problem involves the aorta, which is the main blood vessel distributing oxygenated blood from the heart to all parts of the body through systemic circulation. Heart valves may also be affected.
Types of cardiovascular complications include:
1) Aortic enlargement
The aorta can become dilated, and its walls may bulge, leading to aortic aneurysm. Complications for patients with Marfan syndrome likely start in the section of the aorta closest to the heart.
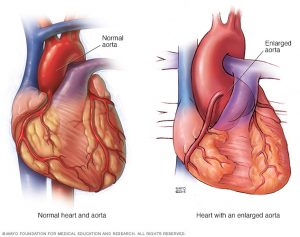
Image showing the difference between a normal heart & aorta, and heart with an enlarged aorta
Image credit: http://www.mayoclinic.org/diseases-conditions/marfan-syndrome/symptoms-causes/dxc-20195415
2) Aortic regurgitation
The aortic valve is situated between the left ventricle and the aorta in the heart. With the aorta becoming enlarged and dilated, there will be leaking of the aortic valve. This causes backflow of blood from the aorta into the left ventricle during ventricular diastole, which results in aortic regurgitation.
3) Mitral valve prolapse
The mitral valve, also known as the bicuspid valve, is a valve in the heart that lies between the left atrium and the left ventricle, allowing blood to flow from the former to the latter. In mitral valve prolapse, damage to the mitral valve tissues can result in the valve sliding backwards into the left atrium during systole, resulting in a late systolic murmur (a murmur as the heart relaxes just before it contracts again).
4) Aortic tear/rupture
A tear in the wall of the aorta causes blood to flow between the layers of the wall. This is also known as aortic dissection, and can lead to aortic rupture and a subsequent decreased blood flow to body organs, causing death.
The signs and symptoms of Marfan syndrome can be apparent at any point from infancy to adulthood, and the severity of the symptoms determine its fatality. It can be challenging to diagnose Marfan syndrome as signs and symptoms vary greatly from person to person. In the majority of cases, a diagnosis of Marfan syndrome is confirmed through a comprehensive evaluation of the person’s family and medical history3. A detailed physical assessment including checking for stretch marks, listening to the heart, and observing physical features (for example, scoliosis and length of fingers/arms) is required for a confirmed diagnosis of Marfan syndrome.
In addition, a diagnostic tool known as Ghent criteria can be used by GPs to differentiate between Marfan syndrome and other similar syndromes. It consists of both major and minor criteria. Examples of ‘major criteria’ include family history, an enlarged aorta, tear in the aorta, at least four skeletal problems, and lens dislocation. Examples of ‘minor criteria’ include stretch marks, loose joints and a high palate. For individuals who have a significant family history of Marfan syndrome, one major and one minor criteria would be sufficient to confirm a diagnosis. For individuals who do not have significant family history, two major and one minor criteria must be observed for a confirmed diagnosis of Marfan syndrome.
While there is currently no cure, patients diagnosed with Marfan syndrome could still have a normal life expectancy after undergoing treatment and appropriate management. Preventative medication for young children is commonly practiced, and arrangement for regular checkups by cardiologists is important to monitor the patient’s aorta and heart valves. Beta-blockers, calcium channel blockers, and ACE inhibitors are common medications used to reduce blood pressure, hence slowing the damage to the heart or blood vessels in Marfan syndrome 4.
Depending on the extent of damage to the aorta and heart valves, surgery may be necessary to repair the damage. Due to the risk of acute aortic dissection, individuals with Marfan syndrome are strongly discouraged from engaging in vigorous sports. However, moderate exercise is strongly encouraged due to its benefits to cardiovascular, skeletal, and psychosocial health in the long run 5.
Progress and advancement in medicine have allowed us to gain a better understanding of the aetiology, pathophysiology, and management/treatment of Marfan syndrome. As medicine continues to improve and advance each day, we anticipate new treatment options that will be able to lengthen and strengthen the quality of life for patients with Marfan syndrome.
Author
References
- The Marfan Foundation. What is Marfan Syndrome [cited 11th July 2016]. Available from: https://www.marfan.org/about/marfan.
- Medscape, Chen H, Descartes M. Genetics of Marfan Syndrome [cited 11th July 2016]. Available from: http://emedicine.medscape.com/article/946315-overview.
- NHS choices. Marfan syndrome [cited 11th July 2016]. Available from: http://www.nhs.uk/conditions/marfan-syndrome/pages/introduction.aspx.
- Genetic Home Reference. Marfan Syndrome: U.S. National Library of Medicine; [cited 11th July 2016]. Available from: https://ghr.nlm.nih.gov/condition/marfan-syndrome – diagnosis.
- Judge DP, Dietz HC. Marfan’s syndrome: 366, No. 9501, p1965-1976 [cited 11th July 2016]. Available from: http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(05)67789-6/abstract.