
AlphaFold: AI Breakthrough for Protein Folding Problem

While the pandemic is still raging, the scientific community has continued to work hard on research in all fields. DeepMind, an artificial intelligence (AI) laboratory based in London, declared in November a milestone achievement – they succeeded in solving the decades old protein-folding problem. One of the most challenging problems in the modern world, the protein folding problem has been unsolved until now.
What is DeepMind and AlphaFold?
DeepMind started in 2010 as a research group focused on application of AI in video games, it made waves in 2016 when its AI program AlphaGo won against a human professional Go player 1. It has since improved upon its algorithms and successfully dominated other games such as chess, Starcraft II, and Atari. AlphaFold is DeepMind’s latest AI program that uses supervised learning techniques to attack the computationally expensive problem of protein folding.
The Protein Folding Problem

Proteins are self-assembling bio-machines on a very small scale, or in technical terms, long molecular chains of amino acids. Haemoglobin, keratin, and insulin are some commonly known proteins to name a few. Scientists are aware of millions of proteins that exist in various biological processes. Although, to put what scientists already know in perspective, the Protein Data Bank (PDB) includes only about 170000 protein structures. 3D shapes of a vast majority of naturally-occurring proteins are still unknown.
These proteins have extremely complex shapes, often looking messy. However, the shapes the proteins take are not haphazard; they are determined by cellular level chemical reactions. The DNA of a species determines the sequence of the amino acids, while the RNA helps transcribe these proteins according to the instructions in the DNA. Molecular biologists refer to this as the central dogma 2. The RNA generates the proteins in a linear fashion, however the proteins do not remain as straight chains for long: they fold and coil up in specific positions. Due to this mechanism, various components of the protein folding in their unique way adds up to create a massive complex 3D structure.
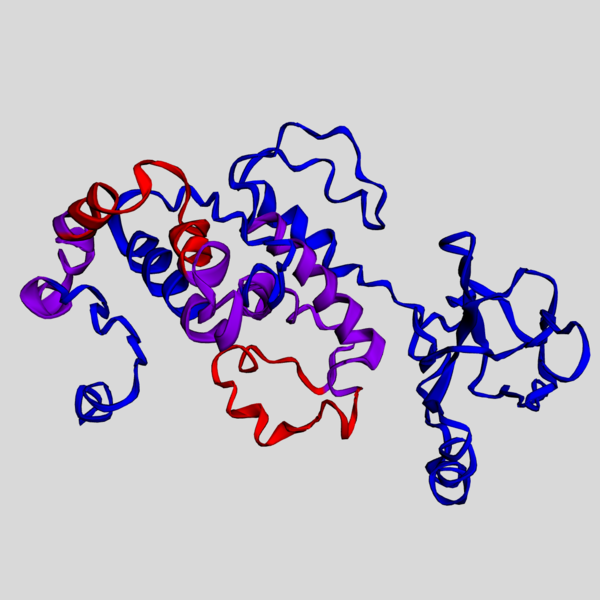
Image credit: Bauma319 on WikiCommons
The 3D shapes of proteins determine which targets they can attach with, analogous to a key fitting only its designated lock. Which also means that the specific shape of proteins is their signature and thus defines their function. For most of the proteins in the PDB, we only know about their amino acid sequence encoded in the DNA, not their structure. If we know the exact shape of a protein and how it is formed we can use this knowledge to target specific protein functions e.g. better treatment of diseases, biological waste treatment, or improving cell performance. Correctly predicting the 3D shape of proteins from the 1D amino acid sequence is the problem of protein folding that has eluded scientists for over half a century.
While X-ray crystallography is a reliable technique to find the protein structures, it is also expensive. Other experimental methods, such as nuclear magnetic resonance (NMR) or electron microscopy, can give clues to protein structures to a certain extent. However, these methods also have limitations. They are cumbersome, expensive, and are not universal. We know protein structures of only a small percentage of existing proteins. Hence, researchers have started exploring alternative ways using the computational power of advanced computers: DeepMind’s AlphaFold is a successful effort in this direction.
A Breakthrough by AlphaFold
Even if computers can be used to do the hard labour, it is no easy task. The number of possible configurations for a given number of atoms in a naturally-occurring protein is astronomical, of the order of 10300 for a standard protein chain molecule 3. It would take millions of years to test each configuration individually. Unleashing the power of artificial intelligence is a way to reduce the prediction time. In 1994, researchers started a biennial competition, Critical Assessment of Structural Prediction competition (CASP) 4 to assess and monitor progress of efforts by comparing the performance of various independent algorithms to correctly predict the known protein structures. It is a unique global platform based on shared knowledge.
The 14th version of CASP (CASP14) was held, mid-2020 in which multiple groups competed to correctly predict the structures of about 90 proteins 5. At the end of the competition, on November 30, 2020, DeepMind researchers announced their breakthrough. The latest version of the program, AlphaFold2 had achieved great success in predicting the structures for the target proteins with the highest median score for Global Distance Test (GDT). In absolute simplest terms, the GDT score (between 0 and 100) is the percentage of the protein structure that is correctly predicted. While the experimental methods are informally considered to have a median score of 90, AlphaFold2 scored 92.4, decidedly higher. While most people expected to reach this level of accuracy in a few decades, DeepMind seems to have not only accelerated the solution at an incredible pace but dominated all other programs competing at CASP14.
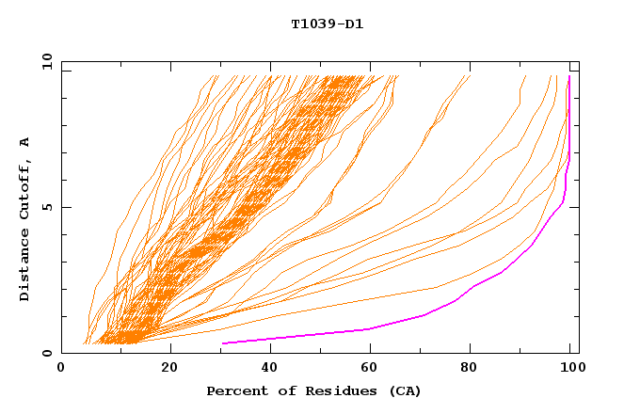
(Image used with permission from Prediction Center and CASP)
AlphaFold2 used multiple deep neural networks for different components of the proteins to predict the optimal distance between a pair of amino acids in the final structure. One of the key aspects of the program that probably made it more accurate is it decides which protein sequence blocks to regard as significant using a numerical confidence measure, discards the rest, and then builds them up for a final global structure which will have a maximum likelihood of being correct. The algorithm has performed really well with a median GDT score of 87 for the most challenging proteins in CASP14.
The results have far-reaching consequences. It gives computational biologists a new tool that is accurate and reliable. Earlier this year, DeepMind used it to predict several protein structures for SARS-CoV-19 virus. Additionally, it is a marker of how powerful AI can become. The future is certainly bright.
This article was specialist edited by Alexander Telfar and copy-edited by Richard Murchie.
Author
References
- https://institutions.newscientist.com/article/2132086-deepminds-ai-beats-worlds-best-go-player-in-latest-face-off/
- https://en.wikipedia.org/wiki/Central_dogma_of_molecular_biology
- https://web.archive.org/web/20110523080407/http://www-miller.ch.cam.ac.uk/levinthal/levinthal.html
- https://en.wikipedia.org/wiki/CASP
- https://www.predictioncenter.org/casp14/index.cgi









{kind=link}