
PARP inhibitors: Synthetic Lethality
Chemotherapy is the traditional drug treatment for cancer, effective yet plagued by its side effects. These agents are toxic to cells (cytotoxic) that rapidly divide, hence killing cancer cells throughout the body. However, there are also healthy cells in our bodies that rapidly grow and divide, such as cells found in our blood, digestive tract and hair follicles. Chemotherapy cannot distinguish between these healthy cells and cancer cells. As a result, the former also becomes damaged during treatment, accounting for the side effects. Though a temporary inconvenience experienced by some patients, chemotherapy can often be harmful and even fatal to others. The lack of specificity of chemotherapy agents is a major driver in cancer treatment research, to discover new agents more selective for cancer cells and therefore less toxic to patients. This is where molecularly targeted therapies come into play.
Molecularly targeted therapies are drugs in cancer treatment that target specific molecules involved in cancer cell growth and progression. The aim is to improve safety in patients as they precisely target cancer cells. Hence, normal or healthy cells evade the effects of these drugs. Poly ADP ribose polymerase (PARP) inhibitors represent a novel class amongst the targeted therapies currently in development.
These drugs work via a mechanism known as ‘synthetic lethality’, distinguishing them from other therapies and representing a new treatment strategy in cancer. Synthetic lethality, a rather ominous term, may conjure images of an exceptionally dangerous drug carefully designed to carry out fatal tasks within those administered it. In theory this is true, since this concept underpins how PARP inhibitors selectively target cancer cells and destroy them. However, this mechanism also underlies the sparing of healthy cells, revealing that the term is not as lethal as it may sound. Furthermore, it provides the basis for the use of PARP inhibitors in subpopulations of cancer patients, representing a class of breakthrough drugs in the modern era of precision medicine.
The term was first coined by Dobzhansky in 1946, describing a situation in which the presence of one of two mutations within a cell has little or no effect but a combination of both is lethal 1. Now the term has been extended to cases where combinations of a mutation and drug induce cell death, but in the presence of either on their own the cell can survive. This concept can easily be pictured in terms of riding a bicycle downhill towards a large traffic junction. As you approach the bottom of the hill you pull on both your right and left brake, safely coming to a stop. If either brake was broken you could probably still manage to stop, though it may prove a bit more difficult. However, if you discovered both brakes were not working, you would go speeding down the hill and likely end up in an accident. This analogy encompasses the concept of synthetic lethality, where in the presence of a mutation or drug a cell can still function, but a combination of both renders the cell non-viable.
The primary treatment strategy in the development of PARP inhibitors has been the exploitation of synthetic lethality by using these drugs in breast and ovarian cancers with BRCA gene mutations. BRCA1 and BRCA2 genes encode for tumor suppressor proteins that help repair double-strand DNA breaks following damage 2. Mutations in BRCA genes lead to defects in their protein products, impairing their ability to fix DNA which in turn leads to mutations. Consequently, these cells become more likely to accumulate mutations over time that may lead to cancer. With the BRCA-mediated DNA repair mechanism being shut down, BRCA-mutated cells become more reliant on an alternative pathway to repair DNA, the PARP pathway.
PARPs are a family of enzymes involved in molecular pathways that also play an important role in the DNA damage response. They repair single-strand DNA (ssDNA) breaks, crucial for preventing double-strand DNA (dsDNA) breaks from forming, since replication of single strand breaks can give rise to double-strand breaks. PARP inhibitors, as the name suggests, block PARPs therefore suppressing this only alternate repair mechanism in BRCA mutated cancer cells. Hence, cancer cells are unable to repair dsDNA breaks, causing cell death. This underlies the pronounced cytotoxicity observed in preclinical studies of PARP inhibitors in tumors with BRCA deficient cells 3.
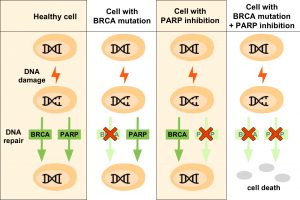
In healthy cells, the PARP pathway and BRCA tumor suppressor proteins are involved in DNA repair following DNA damage. Cells with BRCA mutations become dependent on the PARP pathway to fix damages to DNA, since the protein product of BRCA genes is altered and defective. Cells exposed to PARP inhibitors repair DNA via the protein products encoded by BRCA genes if they become damaged. In cells with BRCA mutations that are treated with PARP inhibitors, both DNA repair mechanisms are blocked and the cell dies.
These findings have translated into promising results from emerging clinical trials. One noteworthy result was the success of an early human trial that led to the accelerated approval of Olaparib in 2014 for treatment of relapsed BRCA-mutated advanced ovarian cancer 4. This was a major landmark for PARP inhibitors and cancer alike, becoming the first drug approved for ovarian cancer since 2003. Trials currently underway are also investigating the potential use of PARP inhibitors (e.g. Olaparib, Veliparib, Niraparib) in other types of cancer with BRCA mutations or BRCA-like features (known as BRCAness) including breast, endometrial, prostate and pancreatic cancer. The use of these drugs in predefined populations of patients highlights a crucial element of PARP inhibitors that is changing the face of cancer treatment.
This development will play a key role in precision medicine, which refers to treating an individual’s disease based on their genetic profile. Each cancer subtype has different genetic variants that affect how the patient will respond to treatment. Precision medicine seeks to ensure patients are taking the drugs which they are likely to respond to and therefore ascertain that benefits outweigh the risks. PARP inhibitors are not the first drugs in development as precision medicine for cancer. Nonetheless, precision medicine is still in its infancy, marking the importance of the success of PARP inhibitors which have been demonstrated so far. In addition to ovarian cancer, Olaparib could soon become the first precision medicine approved for prostate cancer, currently being in the final phase of clinical trials. This highlights another advantage of PARP inhibitors, namely the potential for accelerated drug development. Theoretically, if drugs are matched to patients most likely to respond, trials will be shorter and results will become available sooner, as already seen with the accelerated approval of Olaparib. Overall, this may provide faster access to cancer patients who are likely to benefit from these drugs.
Despite recent findings and the great potential of PARP inhibitors, it should be acknowledged that it still remains unclear whether genetic testing can reliably determine which patients will respond. This highlights the importance of addressing the ethical implications this class of drug carries with it. Foremost, the requirement of genetic testing means subsequent tough decisions must be made based on possibly inaccurate information. What about patients for whom treatment does not work despite the expectation it should? Therefore, it is essential for this prerequisite to establish whether a patient will respond or not, since failure to do so would make it obsolete. In addition, once a genetic profile is established this can raise serious emotional and social issues if treatment is restricted to subpopulations of patients. PARP inhibitors may prove to be of use in other types of cancers with defects in DNA repair, and on-going trials are seeking to provide more clues or answers surround these drugs and resolve some of the aforementioned issues. Thus, while PARP inhibitors represent a promising class of drugs in cancer treatment, exhibiting the unique strategy of synthetic lethality, the extent to which these drugs may prove successful remains to be revealed.
This article was specialist edited by Alisha Aman and copy edited by Rebecca Laidlaw.
Author
References
- http://www.sciencedirect.com/science/article/pii/S0959804910007094
- http://www.sciencedirect.com/science/article/pii/S1574789111000743
- http://www.nature.com/nature/journal/v434/n7035/full/nature03443.html
- http://www.nejm.org/doi/full/10.1056/NEJMoa1105535









